 Најубавата манекенка од 90-тите се соблече и покажа како навистина изгледа")

 Култниот фустан во сенка на прстен: Популарната актерка потврди дека е во брак?")
 Свршеницата на Роналдо повторно е тема на медиумите: во црн фустан ги истакна своите облини")

















Со едукативната кампања „Ги запознаваме ретките болести“ на Здружението за лица со ретки болести „Живот со предизвици“, секој ден во февруари се зборува за една ретка болест, начинот на дијагностицирање, третман и предизвиците со кои се соочуваат луѓето што живеат со неа. Како ја добиваат дијагнозата? Дали има некаков лек? Колку и дали воошто се пристапни и достапни лековите? – се прашањата кои се откриваат во кампањата.
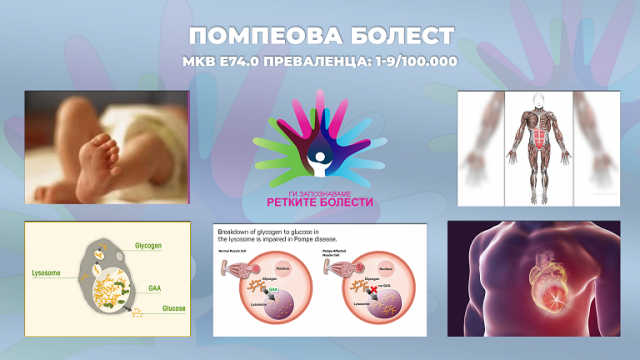
Pompe disease – Помпеова болест
МКВ Е74.0
Преваленца: 1-9 /100.000
Помпеовата болест е ретка, прогресивна и метаболна миопатија со глобална затапеност од 1 на 40.000 породувања. Спаѓа во групата на пореметувања околу складирањето на лизозомите кое е предизвикано од дефицит на лизозомалната алфа-глукозидаза-ензим кој го разградува лизозомалниот гликоген до глукоза. Дефицитот на овој ензим доведува до масивна депозиција на гликогенот во различни ткива, особено срцевата и скелетната мускулатура (вклучувајќи ги и респираторните мускули). Сето тоа доведува до развој на хипертрофична кардиомиопатија и прогресивна мускулна слабост, како и пореметување на респираторната функција.
Ова заболување се наследува автосомно-рецесивно. Болеста е предизвикана од мутација на ген кој се наоѓа на долгиот крак од 17-от хромозом. Повеќето случаи се должат на три мутации. Трансверзијата на тиминот со гванинот како мутација е најчеста кај адултната форма од ова нарушување.
Клиничката презентација на Помпеовата болест може да се опише како спектар на заболувања кој се движи од брзо прогресирачка инфантилна форма до побавно прогресирачка доцна форма.
Кај инфантилната форма на Помпеовото заболување можат да не се појават никакви симптоми при раѓањето. Оваа форма обично се појавува во првите три месеци од животот со брзо прогресивна мускулна слабост, намален тонус на мускулите (хипотонија), респираторна инсуфициенција и еден вид на срцева болест, позната како хипертрофична кардиомиопатија, состојба која се карактеризира со абнормално задебелување на ѕидовите на срцето (главно тоа се левата комора и преградата помеѓу левата и десната комора) што резултира со намалена срцева функција. Овие проблеми заедно кулминираат во кардиореспираторна слабост која се јавува во првите две години од животот. Многу новороденчиња имаат голем јазик и умерено зголемување на црниот дроб. Нозете им се во позиција како кај жаба, а ткивата се цврсти на палпација (псевдо-хипертрофија). Вообичаен е проблемот со хранењето и голтањето, како и проблемот со дишењето, па често завршуваат со инфекции на респираторниот тракт. Моторниот развој е целосно застанат. Менталниот развој е обично нормален. Практично, сите новороденчиња доживуваат губење на слухот. Класичната инфантилна форма на Помпеовата болест се карактеризира со целосен недостаток на активноста на лизозомалната алфа-глукозидаза, а како резултат на тоа, се складира гликогенот во скелетните мускули и срцето.
Доцната форма на Помпеовата болест се манифестира во детството, адолесценцијата или во повозрасните години. Истата е бавно прогресивна отколку инфантилната форма. Пациентите со оваа форма на болеста се карактеризираат со прогресивна миопатија, предоминација на проксималните мускули на карлицата и рамењата, како и различни степени на респираторно пореметување со што пациентот се доведува до потреба од респираторна помош. Временскиот интервал за кој болеста прогресира е варијабилен и непредвидлив. Кај некои пациенти се забележува брзо пореметување на функцијата на респираторните и скелетните мускули што доведува до откажување на дишењето додека кај други пациенти, прогресијата е побавна. Кај друга група пациенти пак, се забележува дисоцијација во прогресијата на скелетната и срцевата мускулатура.
Дијагностика:
Дијагнозата на Помпеовата болест се заснова на темелна клиничка процена, детална лична анамнеза, како и детална семејна анамнеза и сите се истражуваат со најразлични дијагностички тестови. Пренаталната дијагностика е можна само за онаа бременост за која се смета дека постои ризик за развој на Помпеова болест.
При инфантилната рана форма, кај новороденчето се забележува слабо хранење, неуспешен развој, како и компликации со дишењето. Типичните наоди вклучуваат зголемено срце со неспецифични дефекти во спроводливоста на срцевата активност. Биохемиските испитувања вклучуваат зголемување на серумската креатин-киназа (обично околу 10 пати) пропратена со поблаги покачувања на серумската алдолаза, аспартатна трансаминаза, аланин трансаминаза и млечна дехидрогеназа. Дијагнозата се потврдува со евалуација (процена) на активноста на лизозомалната алфа-глукозидаза преку земање на примерок од крвта и генетско тестирање.
При доцната форма, кај возрасното лице постепено ќе се појавува прогресивна слабост на рацете и нозете, со влошување на респираторната функција. Електромиографијата може да се користи за да ги исклучи останатите причини за слабост на екстремитетите. Наодите при биохемиските тестови се слични како оние кај инфантилната форма, со тоа што треба да се има во предвид фактот дека креатинин-киназата може да биде во нормални граници кај некои случаи. Дијагнозата се поставува со процена на ензимската активност преку земање на примерок од крвта и генетско тестирање.
Третман:
Срцевите и респираторните компликации се третираат симптоматски. Физикалната терапија може да биде корисна за некои пациенти. Измените во исхраната можат да обезбедат привремено подобрување, но не го менуваат текот на болеста. Генетското советување на семејствата може да им обезбеди информации за ризикот од заболување во текот на следните бремености.
На 28 април 2006 година, Американската Агенција за храна и лекови (ФДА) ја одобри апликацијата за биолошка лиценца за Миозимот (ензимска супституциона терапија). Воедно, миозимот е првиот третман за пациентите со ова заболување. Раната дијагностика и раниот третман доведуваат до многу подобар исход. Треба да се напомене дека можни се и несакани ефекти како што се: треска, црвенило, осип по кожата, тахикардија, па дури и шок.
На 14-ти август 2006 година, Канада го одобри Миозимот за третман на пациентите со Помпеова болест. Подоцна миозимот добива широко одобрување и во Европската Унија.
На 26-ти мај 2010 година, ФДА го одобри и Лумизимот, слична верзија на Миозим, за третман на зрелата форма на Помпеовата болест. Лумизимот и Миозимот имаат иста генеричка состојка (алфа-глукозидаза). Разликата помеѓу овие два производи е во процесот на производство
Прогноза:
Прогнозата за лицата со Помпеово заболување варира во зависност од почетокот и тежината на симптомите. Без третман, болеста е смртоносна, особено кај новороденчињата и малите деца. Миозимот е рекомбинантна форма од хуманата форма на лизозомалната алфа-глукозидаза, која во моментов се користи како замена за исчезнатиот ензим. Студиите покажуваат дека третманот со Миозимот јасно го продолжува целокупното преживување кај пациентите со инфантилна форма на Помпеовата болест. Покрај тоа, студијата покажа дека започнувањето на третманот пред 6-месечна возраст доведува до намалување на смртноста и инвалидитетот поврзани со ова погубно нарушување. Тајван и САД го започнаа скринингот за новороденчиња. Резултатите од раната дијагностика и раната иницијализација на терапијата, драматично го подобруваат исходот на болеста. Многу од овие новороденчиња подоцна имаат нормален моторен развој.
За проектот:
Гордана Лолеска, Супер радио и „Живот со предизвици“ и овој февруари ве запознаваат со ретките болести во Република Македонија со надеж дека преку овие 28 ретки дијагнози сите заедно ќе научиме нешто повеќе.
Првиот ден на ретки болести беше одбележан во 2008 година, точно на 29 февруари, „редок“ датум кој се случува само еднаш на секои четири години. Од тогаш, Денот на ретки болести се одржува секоја година на последниот ден од февруари, месец познат по тоа што има „редок“ број на денови.
Денот на ретките болести е меѓународна кампања за подигнување на свеста за ретките болести. Од почетокот во 2008 година, илјадници настани се организираат низ целиот свет и опфаќаат милиони луѓе. Кампањата започна како европски настан и со текот на времето стана глобален феномен, со учество на над 90 земји од целиот свет во 2018 година.
Република Македонија за прв пат се вклучи во одбележување на овиј редок ден во 2013 година.
Кампањата за Денот на ретки болести е за пошироката јавност, за релевантните институции и секој е добредојден да се придружи: пациенти и семејства, организации на пациенти, медицински професионалци, истражувачи, фармацевтска индустрија, јавни здравствени институции – колку повеќе не има, толку подобро. Денот на ретките болести е можност за учесниците да бидат дел од глобалниот повик кон креаторите на политики, истражувачите, компаниите и здравствените професионалци повеќе и поефикасно да ги вклучат пациентите во истражувањата за ретки болести.
Republika.mk – содржините, графичките и техничките решенија се заштитени со издавачки и авторски права (copyright). Крадењето на авторски текстови е казниво со закон. Дозволено е делумно превземање на авторски содржини (текст и фотографии) со ставање хиперлинк до содржината што се цитира.


